北京医疗器械设计开发技术服务
与我国法规设计开发投入要求相比,美国FDA医疗器械法规的设计开发要求则更为具体。美国FDA于1997年发布的设计开发内容在《医疗器械质量体系规范》(FDA21CFR820)中纳入医疗器械质量管理体系法规,要求制造商建立和维护质量管理体系涵盖从设计、开发到服务的整个过程。其中包括:制造商应建立和维护程序,应确保与设备相关的设计要求是适当的,讨论设备的预期用途,包括用户和患者的需求,并在请求输入时建立机制以解决不完整、模棱两可等问题,要求、输入应形成文件并审查和批准等。好的医疗器械设计应该考虑到使用者的舒适感和便捷性。北京医疗器械设计开发技术服务

医疗器械设计开发的流程主要包含策划、设计输入、设计、设计验证、设计确认、设计转换、上市准备、上市监控整个产品生命周期。从设计输入部分来说,产品需求规范需要满足多方面的要求,例如用户需求、法规标准、风险管理、竞品、专利分析、同类产品信息、预期用途、适应禁忌、规格、尺寸外观、生物学性能、物理性能、化学性能、灭菌、可靠性、可用性、追溯性、兼容性要求、包装、标识、运输与存储等。各种标准和要求确实会对新入行的企业造成一定困扰。那么缺乏经验的医械企业该如何有效规避设计开发过程中的风险呢?医疗器械设计开发主文档医疗器械设计流程中,验证产品功能和性能至关重要。

YY/T0287-2017(ISO13485:2016,IDT)《医疗器械质量管理体系用于法规的要求》规定了设计和开发的输入内容,完善了YY/T 0287-2003。设计输入应包括:根据预期用途的功能、性能和安全要求;适用法律法规的要求;适用时,以前类似设计提供的信息;设计和开发所需的其他要求;风险管理的输出;评审输入以确保输入是充分和适当的,要求应该是完整的、清晰的和不矛盾的。在医疗器械质量体系法律法规中,我国2000年发布的《医疗器械生产企业质量体系考核办法》规定了设计开发、产品检验等过程中的质量管理要求。2014年12月发布的《医疗器械生产质量管理规范(试行)》修订了《医疗器械生产质量管理规范》的相关内容后,要求设计和开发的输入应当包括针对预期用途规定的功能、性能和安全性要求;监管要求;风险管理控制措施和其他要求;设计和开发输入应得到评审和批准,并保持相关记录。
思脉得医疗科技公司一直致力于为客户提供高质量的客户服务。我们的客户服务团队可以随时回答客户的问题和需求,提供技术支持和咨询服务。我们的客户服务团队可以帮助客户解决在产品开发过程中遇到的各种问题,确保客户的满意度和产品的质量。总之,思脉得作为一家专业的医疗器械设计开发服务提供商,可以为客户提供完整的设计开发服务,包括需求分析、概念设计、详细设计、样机制作、验证测试、产品认证等多个环节。我们拥有专业的团队和强大的创新能力,注重产品合规性和客户服务,可以为客户提供质量、可靠的设计开发服务。CDMO服务模式可以使医疗器械企业更加灵活应对市场变化。
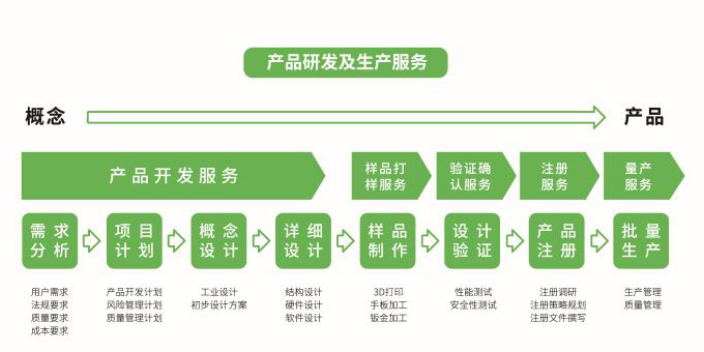
医疗器械设计开发变更相关内容分享:设计和开发变更可能发生在任何阶段,例如:在设计开发审查、验证、确认、设计转换过程中发现的问题而导致的变更;风险管理活动所需的变更;针对上市后发生问题的纠正和预防措施;外部因素的变化(技术发展、法规或技术标准、客户要求)等。会引起变更的因素有很多,例如:设计阶段的遗漏或错误(如计算、材料选择);制造、安装、服务、人体工程学多方面的问题等。设计变更需要以适当的方式进行审查、验证和确认,以确保风险可控。企业可以将设计开发外包给专业服务商,减少资源浪费,提高效率。医疗器械OEM加工供应
思脉得医疗科技集团以创新技术与服务为宗旨,为客户提供超预期的价值。北京医疗器械设计开发技术服务
《医疗器械监督管理条例》钟明确指出,医疗器械监管的主要目的之一就是保证医疗器械的安全有效。《医疗器械注册备案管理办法》第三条规定,医疗器械注册是对提交注册的医疗器械的安全性、有效性和质量可控性进行审查,并决定是否批准申请的过程。设计是实现产品安全性和有效性的重要环节之一。在医疗器械产品的整个生命周期中,设计和开发是产品质量的基础,产品注册信息是设计和开发活动成果的一部分。设计和开发的输入基本上可以视为设计和开发过程中极其重要的环节,是后续设计和开发活动的基础,是识别和定义需求的过程,对注册申请资料的符合性有重要影响。北京医疗器械设计开发技术服务
上一篇: 北京医疗器械设计开发类型
下一篇: 北京医疗器械设计开发开发服务